Tunnetuimmat epätyypilliset periytymistavat
Epätyypilliset periytymistavat ovat nimestään huolimatta osa normaaleja periytymismekanismeja.
Epätyypilliset periytymistavat eivät noudata periytymisen jo kauan tunnettuja pääperiaatteita ja sääntöjä. (Tiivistelmän periytymisen pääperiaatteista löydät Periytymisen periaatteet -osiosta taulukosta 5). Epätyypilliset periytymistavat ovat osa normaaleja biologisia mekanismeja, jotka kohdistuvat DNA:han ja vaikuttavat siihen, millaisia olemme.
Epätavalliset periytymistavat ovat lääketieteellisesti tärkeitä: ne voivat vaikuttaa joidenkin tiettyjen sairauksien ja oireyhtymien ilmenemiseen ja oirekuvaan. Usein epätyypilliset periytymistavat tuovat haasteita diagnostiikkaan ja perinnöllisyystodennäköisyyksien ennustamiseen seuraavissa sukupolvissa. Epätyypilliset periytymistavat on otettava huomioon mm. perinnöllisyysneuvonnassa.
Tunnetuimmat epätyypilliset periytymistavat liittyvät mitokondrioiden DNA:han, Y-kromosomiin ja tietyllä tapaa leimattujen geenien periytymiseen. Muita tärkeitä epätyypillisiä periytymisen mekanismeja ovat toistojaksomuutokset (niin kutsutut dynaamiset mutaatiot) ja X-kromosomin vinoutunut inaktivaatio.
1. Mitä on mitokondriaalinen DNA?
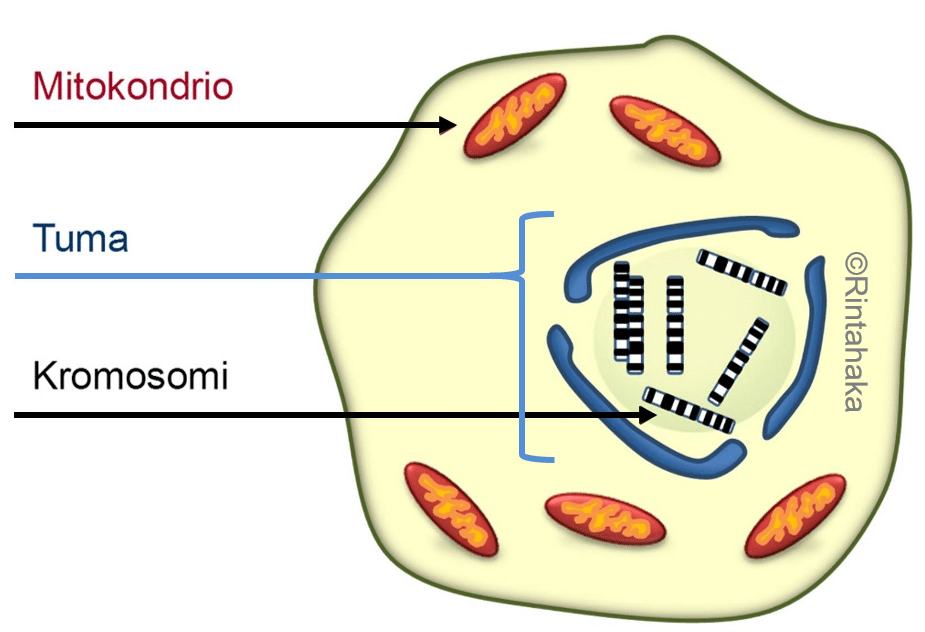
Tumassa olevien kromosomien lisäksi perimäainesta eli DNA:ta on myös mitokondrioissa, jotka ovat solujen energia- ja aineenvaihdunnalle elintärkeitä soluelimiä. Mitokondriaalinen DNA käsittää noin prosentin (1 %) perimämme kokonaismäärästä (16 569 emäsparia vs. 3 609 003 417 emäsparia).
Mitokondrioiden lukumäärän on pysyttävä vakiona solusukupolvesta toiseen. Samalla myös niiden sisältämän mitokondriaalisen DNA:n on monistuttava ja pysyttävä mahdollisimman muuttumattomana. Emosolun jakautuessa, myös sen sisältämät mitokondriot jaetaan tytärsolujen kesken. Itse mitokondriot lisääntyvät jakautumalla, samantapaisesti kuin emosolu jakautuu kahdeksi tytärsoluksi. Tätä edeltää mitokondrioiden perimäaineksen kopioiminen tytärmitokondrioita varten. Tämä prosessi on altis uusille mutaatioille.
1.1 Miten mitokondriaalinen DNA periytyy?
Kehittyvä yksiö perii mitokondriot ja niiden sisältämän mitokondriaalisen DNA:n lähes poikkeuksetta vain äidiltään. Tämä johtuu siitä, että siittiössä on huomattavasti vähemmän mitokondrioita kuin kypsässä munasolussa (noin 50–75 kpl vs. 100 000 kpl). Näin ollen siittiön mitokondriot jäävät lukumäärällisesti vähemmistöön hedelmöittyneessä munasolussa, tai niitä ei päädy hedelmöityksen yhteydessä lainkaan munasoluun. Lisäksi on havaittu, että hedelmöityksessä munasoluun mahdollisesti päätyneet siittiöiden mitokondriot häviävät toistuvien solunjakautumisten aikana alkion kehityksen edetessä. Ehkä äidiltä perityt mitokondriot jakautuvat soluissa vilkkaammin ja niiden lukumäärä täten moninkertaistuu suhteessa isältä perittyihin mitokondrioihin.
1.2 Milloin mitokondrion DNA:ssa oleva geneettinen muutos johtaa sairauteen?
Mitokondrion DNA on herkempi DNA:n emäsjärjestyksen muutoksille kuin tuman kromosomaalinen DNA. Osa mitokondrion DNA:ssa tapahtuneista muutoksista saattaa häiritä mitokondrion geeneistä valmistuvien geenituotteiden toimintaa niin, että ne altistavat yksilön mitokondriosairaudelle. Mitä enemmän yksilössä on mitokondrioita, joiden DNA:ssa on tietty patogeeninen eli sairautta aiheuttava mutaatio, sitä todennäköisemmin henkilö sairastuu johonkin mitokondriosairauteen. Tällainen mitokondriosairauteen johtava geneettinen muutos voi olla a) peritty äidin mitokondrioista tai b) se on uusi ja ainutlaatuinen, niin kutsuttu de novo-muutos, joka on syntynyt sattumalta mitokondrion perimään.
De novo-muutoksista voit lukea kohdasta DNA:n emäsjärjestyksen muutokset.
Mitokondrion DNA:n muutoksista johtuvia sairauksia/oireyhtymiä tunnetaan useita kymmeniä. Esimerkkejä niistä ovat mm. Leighin oireyhtymä ja Kearns-Sayren oireyhtymä. Osa mitokondriosairauksista todetaan jo ennen syntymää ja niiden oireet ovat vakavia. Osa mitokondriotaudeista puhkeaa myöhemmin ja ovat lieväoireisempia. Kaikkein vaikein oirekuva on usein seurausta niistä patogeenisista geenimuutoksista, jotka vaarantavat merkittävästi mitokondrioiden energiantuotantoa tai muuta aineenvaihduntaa. Aivoissa, luustolihaksissa ja maksassa on paljon mitokondrioita, koska niiden energiantarve on suurta ja aineenvaihdunta vilkasta. Usein mitokondriosairauksien oireet ilmenevätkin näissä kudoksissa.
1.3 Miten mitokondrion DNA:sta johtuvat sairaudet periytyvät?
Käytännössä isän mitokondrion DNA:ssa olevat mahdolliset patogeeniset muutokset eivät periydy hänen lapsilleen, sillä kehittyvässä alkiossa on eniten äidin mitokondrioita. Niinpä mitokondrioiden DNA:ssa olevasta muutoksesta johtuvat sairaudet peritään äidiltä. Joskus äiti on lieväoireinen tai oireeton, eikä tiedä mitokondriossa olevasta tautia aiheuttavasta geenimuutoksestaan ennen kuin perheeseen syntyy lapsi, jolla on mitokondriosairaus. (Huom. osa mitokondriosairauksista johtuu kromosomaalisen DNA:n eli tumassa olevan perimäaineksen muutoksesta. Tällöin ne noudattava perinteisiä periytymistapoja. ks. alla)
Mitokondriaalisen DNA:ssa olevien muutosten mahdolliset periytymistavat:
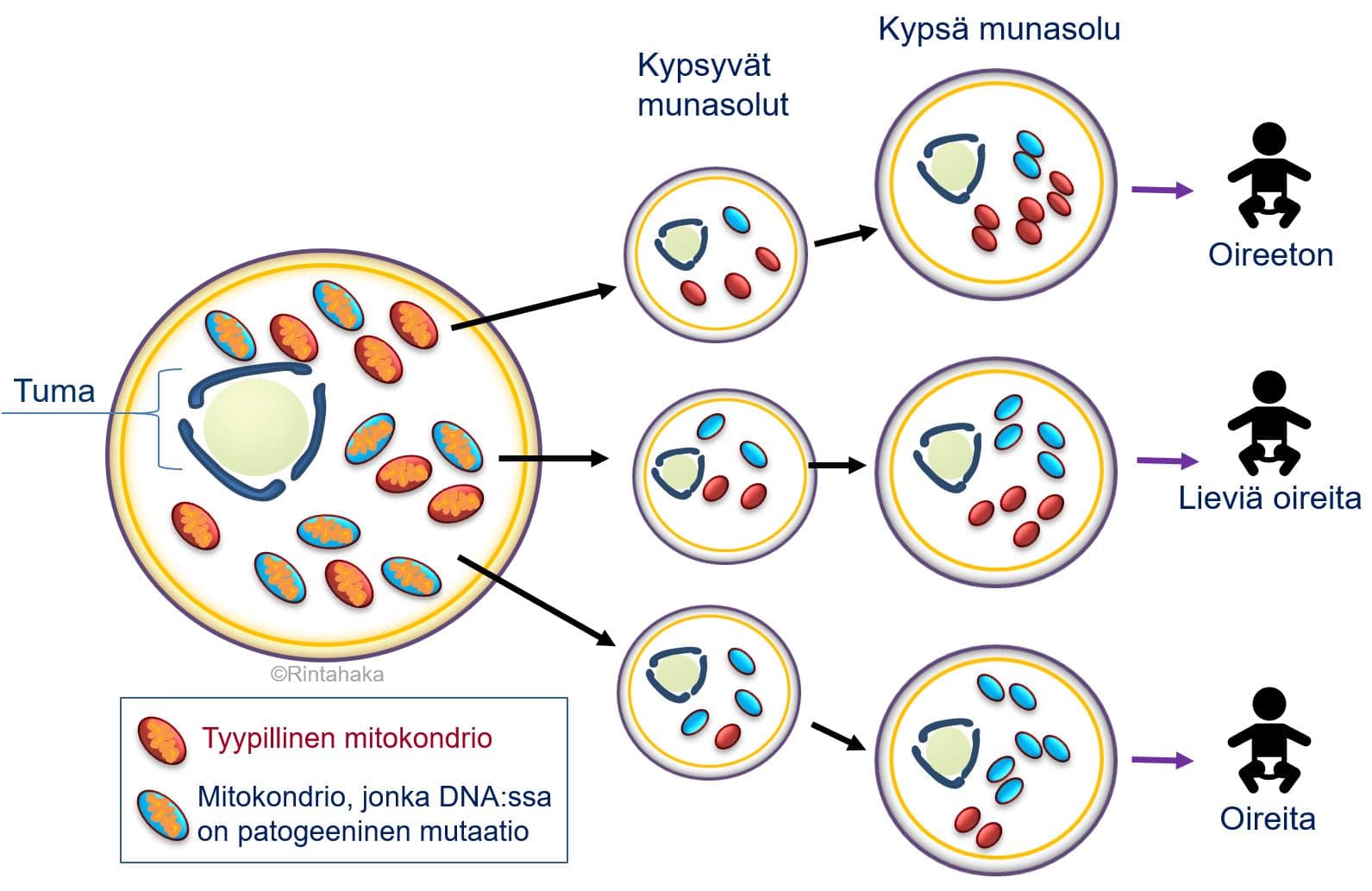
a) Äiti, joka sairastaa mitokondrion DNA:n muutoksesta johtuvaa sairautta, voi siirtää mitokondriosairautensa lapselleen, jos alkioon päätyy runsaasti mitokondrioita, joiden DNA:ssa on tätä sairautta aiheuttavia muutoksia. Toisaalta on myös mahdollista, että äiti, jolla on mitokondriosairaus, synnyttääkin terveen lapsen. Tällöin hedelmöitykseen osallistuneeseen munasoluun on päätynyt enemmän niitä mitokondrioita, joiden perimässä ei ole patogeenisiä muutoksia.
b) Äiti, jolla ei ole todettu mitokondrion perimästä johtuvaa sairautta, voi kuitenkin saada myös lapsen, jolla on mitokondriosairaus. Tässä tapauksessa munasoluun on päätynyt runsaasti mitokondrioita, joissa on patogeeninen muutos siitäkin huolimatta, että ne ovat olleet vain pieni vähemmistö äidin omia mitokondrioita. On myös mahdollista, että patogeeninen muutos on ollut ainutlaatuinen ja uusi eli de novo-muutos. Ratkaisevaa molemmissa tapauksissa a) ja b) siis on, kuinka paljon hedelmöittyneessä munasolussa tai varhaisvaiheen alkiossa on mitokondrioita, joiden DNA:ssa on patogeenisiä muutoksia ja kuinka nämä mitokondriot lukumäärällisesti lisääntyvät eri puolille kehittyvän ja kasvavan yksilön kehoa.
Mitokondriosairaudet voivat aiheutua myös tuman geeneistä olevista muutoksista. Tämä johtuu siitä, että tuman geenit koodaavat runsaasti mitokondrioiden toimintaan ja rakenteeseen tarvittavia geenituotteita, joilla on suuri merkitys mm. elimistön energiantuotannolle ja lukuisille aineenvaihduntareaktioille. Mitokondriosairaudet, jotka liittyvät kromosomaalisen DNA:n geeneihin, periytyvät tavanomaisten periytymissääntöjen mukaisesti. Useimmat näistä mitokondriosairauksista periytyvät resessiivisesti eli peittyvästi.
2. Mitä on geneettinen leimautuminen?
Yleensä sekä isältä että äidiltä perittyjä geenejä käytetään yhtä paljon molemmista vastinkromosomeista (50 % ja 50 %). Perimässä on kuitenkin pieni joukko geenejä, noin 100, joista käytetään normaalisti vain toiselta vanhemmalta (esim. isältä), perittyä geeniä. Samanaikaisesti toiselta vanhemmalta (esim. äidiltä) peritty geeni inaktivoituu tai hiljentyy eli sitä ei käytetä. Tämä tapahtuu geneettisten leimojen, tietylaisten kemiallisten yhdisteiden, avulla. Nämä geneettiset leimat kiinnittyvät DNA:n rakenteeseen niin, etteivät ne muuta DNA:n emäsjärjestystä eli sekvenssiä. Ilmiöstä käytetään nimitystä geneettinen leimautuminen (engl. genomic imprinting). Leimautumisen kautta säädeltävät geenit osallistuvat erityisesti alkion ja sikiön kasvun ja kehityksen säätelyyn.
2.1 Milloin ja miten geenejä leimataan?
Leimautuminen tapahtuu sukusoluissa sukusolujen kehityksen aikana. Tällöin tietyt entsyymit liittävät kemiallisia ryhmiä pieneen osaan munasolun ja siittiön geenejä, niiden emäsjärjestyksen ulkopuolisille säätelyalueille. Munasolussa noudatetaan näiden tiettyjen geenien osalta äidin leimautumisperiaatteita ja siittiössä isän leimautumissääntöjä. Kun sukusolut yhdistyvät hedelmöityksessä, isältä ja äidiltä perityt tietyllä tapaa leimatut geenit alkavat vaikuttaa yhdessä kehittyvässä alkiossa. Toisista geeneistä luetaan siis isän versiota, toisista äidin vastinetta. Tätä tapahtumaa siis ohjataan geneettisillä leimoilla.
2.2 Mikä merkitys leimatuilla geeneillä on syntyvälle lapselle?
Hedelmöityksen tapahduttua isältä ja äidiltä sukusoluissa perityt kemialliset leimat usein säilyvät kehittyvässä alkiossa ja ne myös monistuvat kaikkiin yksilön uusiin soluihin, paitsi hänen sukusoluihinsa. Koska geneettinen leimautuminen jättää aktiiviseksi toisen vanhemman vastingeenin, jokin patogeeninen muutos tässä aktiiviseksi jääneessä geenissä, voi johtaa perinnölliseen sairauteen tai oireyhtymään. Jos tätä aktiivista vastingeeniä ei ole esimerkiksi geenin deleetion eli häviään vuoksi, ja jäljellä on vain toisen vanhemman inaktiivinen vastingeeni, lapselle kehittyy usein oireita/sairaus. Perimäntutkimuksissa geenin emäsjärjestyksen eli sekvenssin tutkiminen ei siis riitä kertomaan käytetäänkö kyseistä geeniä aktiivisesti geenituotteiden valmistamiseksi. Tähän tarvitaan geenien leimautumista tutkivaa epigenetiikkaa, joka siis perustuu sekvenssin ulkopuolisten alueiden kemiallisten leimojen tutkimiseen. (”Epi” etuliite tulee muinaiskreikasta ja se tarkoittaa ”päällä” tai ”yläpuolella”.)
Muutokset sukupuolisidonnaisesti leimatuissa geeneissä tai leimautumisesta vastaavissa geeneissä (niin kutsutuissa leimauskeskuksissa), voivat siis edesauttaa tiettyjen resessiivisesti eli peittyvästi periytyvien sairauksien ilmenemistä. Aiheesta lisää alla.
2.3 Mitä voi tapahtua, jos geeni on poikkeavasti leimautunut tai se puuttuu?
Jos yksilön geenien ja/tai niihin liittyvien DNA-alueiden leimautuminen on jostakin syystä häiriintynyt, seurauksena voi olla erilaisia oirekokonaisuuksia, toistuvia keskenmenoja tai hedelmättömyyttä.
Esimerkiksi kromosomissa-15 ja -11 on alueita, joiden poikkeava leimautuminen voi johtaa tiettyihin kehitysvammaoireyhtymiin. Tavallisesti kromosomissa-15 sijaitsevaa UBE3A-nimistä geeniä luetaan aivoissa vain äidiltä peritystä kromosomista-15. Isän UBE3A-geeni on inaktiivinen eli sitä ei käytetä geenituotteiden valmistamiseksi. Jos lapsella on äidiltä perityssä kromosomissa-15 deleetio eli häviämä, joka poistaa UBE3A-geenin, seurauksena tästä on Angelmanin oireyhtymä.
Joskus geenin leimautuminen on epätavanomainen. Tämä voi johtua niin kutsutusta leimauskeskusmuutoksesta. Leimauskeskuksen muutokset ovat erittäin harvinaisia, mutta silti mahdollisia. Niiden yleisyydeksi esimerkiksi kaikista Angelmanin ja Prader-Willin oireyhtymätapauksista on arvioitu olevan alle 5 %:n luokkaa.
Tarkemmin kromosomialueiden-15 ja -11 leimautumisesta, niihin liittyvistä kehitysvammaoireyhtymistä ja niiden perinnöllisyydestä voit lukea lisää Harvinaiskeskus Norion sivuilta: Angelmanin oireyhtymä, Prader-Willin oireyhtymä, Russel-Silverin oireyhtymä ja Beckwith-Wiedemannin oireyhtymä.
2.4 Periytyykö tietty geneettinen leimautumistapa seuraavalle sukupolvelle?
Geneettinen leimautuminen puretaan ja uudelleen ohjataan kehittyvän yksilön sukusolujen kantasoluissa sekä hedelmöityksen ja varhaisen alkionkehityksen aikana. Ihmisillä sukusolujen geneettisten leimojen periytymistä on vaikea tutkia. Lisäksi ihmisellä on useita erilaisia epigeneettisiä säätelyjärjestelmiä, jotka voivat vaikuttaa geenien leimautumiseen ja aktiivisuuteen. Toistaiseksi voidaan varmuudella sanoa, että geneettinen muutos leimauskeskuksessa tai geenissä, jonka alueeseen geneettinen leima tavanomaisesti liitettäisiin, voivat johtaa oireisiin ja että nämä muutokset voivat periytyä.
Perimää ja perinnöllisyyttä tarkasteltaessa on siis aina huomioitava, ettei DNA:n emäsjärjestys ole ainut tekijä, joka ohjaa kehitystä ja ominaisuuksiamme. Myös epigenetiikan eli leimautumisen tarkastelu on joissakin tilanteissa välttämätöntä.
3. Mitä ovat toistojaksomuutokset eli dynaamiset mutaatiot?
Ihmisen normaaliin perimään kuuluu monia peräkkäisiä kolmen emäksen jaksoja, jotka toistuvat lukuisia kertoja DNA:ssa. Kun näiden toistojaksojen lukumäärä ylittää tietyn kynnysarvon, kyseinen perimän alue muuttuu epävakaaksi. Epävakaita toistojaksoja kutsutaan esimutaatioiksi. Ne voivat laajeta edelleen, josta seurauksena voi olla täysimittainen toistojaksomuutos ja sen aiheuttama sairaus tai oireyhtymä.
3.1 Miten toistojaksomuutokset periytyvät ja voivatko ne aiheuttaa oireita?
Lapsi voi periä isänsä tai äitinsä epävakaan toistojaksomuutoksen. On vaikea ennustaa, kuinka vanhemman toistojaksomuutos käyttäytyy lapsen perimässä, koska ne ovat dynaamisia, eikä niiden käyttäytymistä voida etukäteen ennustaa. Täten on vaikea myös arvioida millaisia oireita lapsella mahdollisesti on ja mitkä niiden vaikeusasteet ovat.
Esimerkkejä toistojaksosairauksista tai -oireyhtymistä ovat mm. fragiili X-oireyhtymä, Huntingtonin tauti ja Myotoninen dystrofia tyypit 1 ja 2. Valtaosa kaikista toistojaksosairauksista periytyy autosomissa dominoivasti eli vallitsevasti, mutta esimerkiksi fragiili X-oireyhtymä periytyy X-kromosomissa resessiivisesti eli peittyvästi.
4. Mitä tarkoittaa X-kromosomin vinoutunut inaktivaatio?
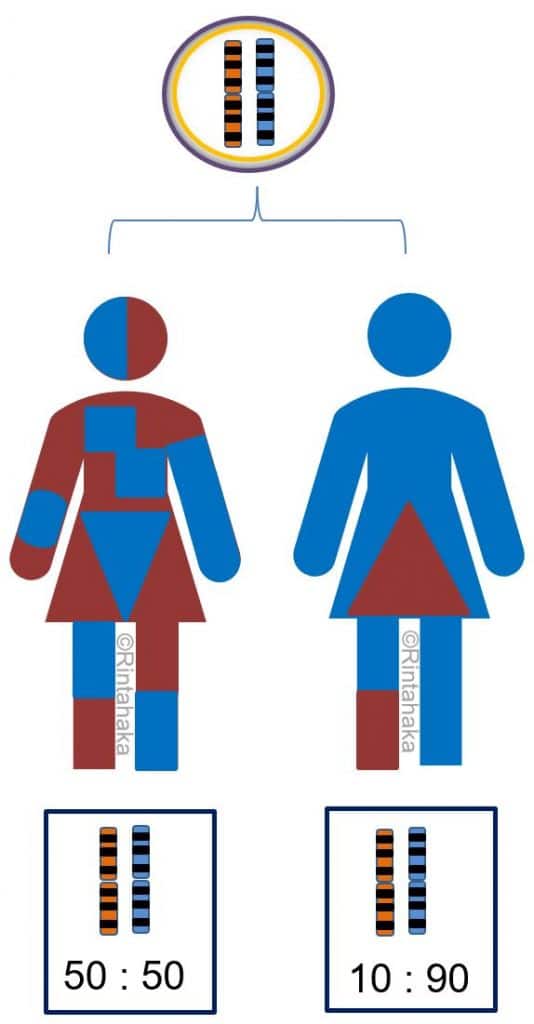
X-kromosomin inaktivaatio tapahtuu vain tytöillä/naisilla. Siinä naisen kahdesta X-kromosomista vain toista käytetään geenituotteiden valmistamiseen ja toinen X-kromosomi ”hiljentyy” tai inaktivoituu eli sitä ei käytetä geenituotteiden valmistukseen. Useimmiten X-kromosomin inaktivaatio on tasapuolista: isältä ja äidiltä perityistä X-kromosomeistä puolet ovat inaktiivisia ja puolet jää aktiivisiksi (50 % ja 50 %). X-kromosomin inaktivaatio takaa, että molemmilla sukupuolilla on käytössään yhtäläinen määrä X-kromosomaalisia geenejä. Miehellähän on tavallisesti perimässään vain yksi X-kromosomi ja naisella kaksi X-kromosomia. (ks. Kromosomit-osio; kuva 5.)
Vinoutunut X-kromosomin inaktivaatio (engl. X-chromosome inactivation skewing, XCI skewing) tarkoittaa tilannetta, jossa toista X-kromosomia käytetäänkin geenituotteiden valmistamiseen merkittävästi enemmän kuin toista. Esimerkiksi isältä perittyä X-kromosomia saatetaankin käyttää 90 %:sesti ja äidiltä perittyä vain 10 %:sesti.
X-kromosomin vinoutunut inaktivaatio voi olla solu- ja kudosspesifistä sekä ilmetä tietyssä kehitysvaiheessa tai iässä. Ilmiön vaikutukset riippuvat myös siitä, miten merkittävään geeniin ja kudokseen vinoutunut X-kromosomin inaktivaatio on kohdistunut ja seuraako siitä soluille valintapaineita tai jokin kasvuetu.
4.1 Miten X-kromosomin vinoutunut inaktivaatio voi johtaa oireisiin?
X-kromosomin vinoutunut inaktivaatio voi johtaa tytöllä/naisella oireisiin, jos tietyssä kohdekudoksessa aktiiviseksi jääkin X-kromosomi, jossa on patogeeninen geenimuutos. Tällöin soluun ei tuoteta riittävästi biologisesti aktiivista ja toimivaa geenituotetta, joka olisi kohdekudokselle elintärkeä. Tämä heijastuu tytössä/naisessa erilaisina oireina.
X-kromosomin vinoutunutta inaktivaatiota on havaittu mm. Rettin-oireyhtymässä, Klinefelterin oireyhtymässä, autismissa ja joissakin syövissä ja autoimmuunisairauksissa. Miten X-kromosomin vinoutunut inaktivaatio johtaa kussakin sairaudessa/oireyhtymässä oireisiin, on osin epäselvää.
5. Mitä on uniparentaalinen disomia, UPD?
Uniparentaalinen disomia (UPD) on harvinainen kromosomimuutos, jossa tietyt vastinkromosomit ovatkin peräisin samalta vanhemmalta (isältä tai äidiltä), eikä molemmilta vanhemmilta, kuten normaalisti. UPD syntytapoja on useita (ks. kuva 12 alla). UPD voi johtua trisomian purkautumisesta (engl. trisomic rescue) tai disomisten sukusolujen kohtaamisesta hedelmöityksessä. Disomialla tarkoitetaan tilannetta, jossa sukusolussa on yhden vastinkromosomin sijaan poikkeuksellisesti kaksi jotakin tiettyä kromosomia.
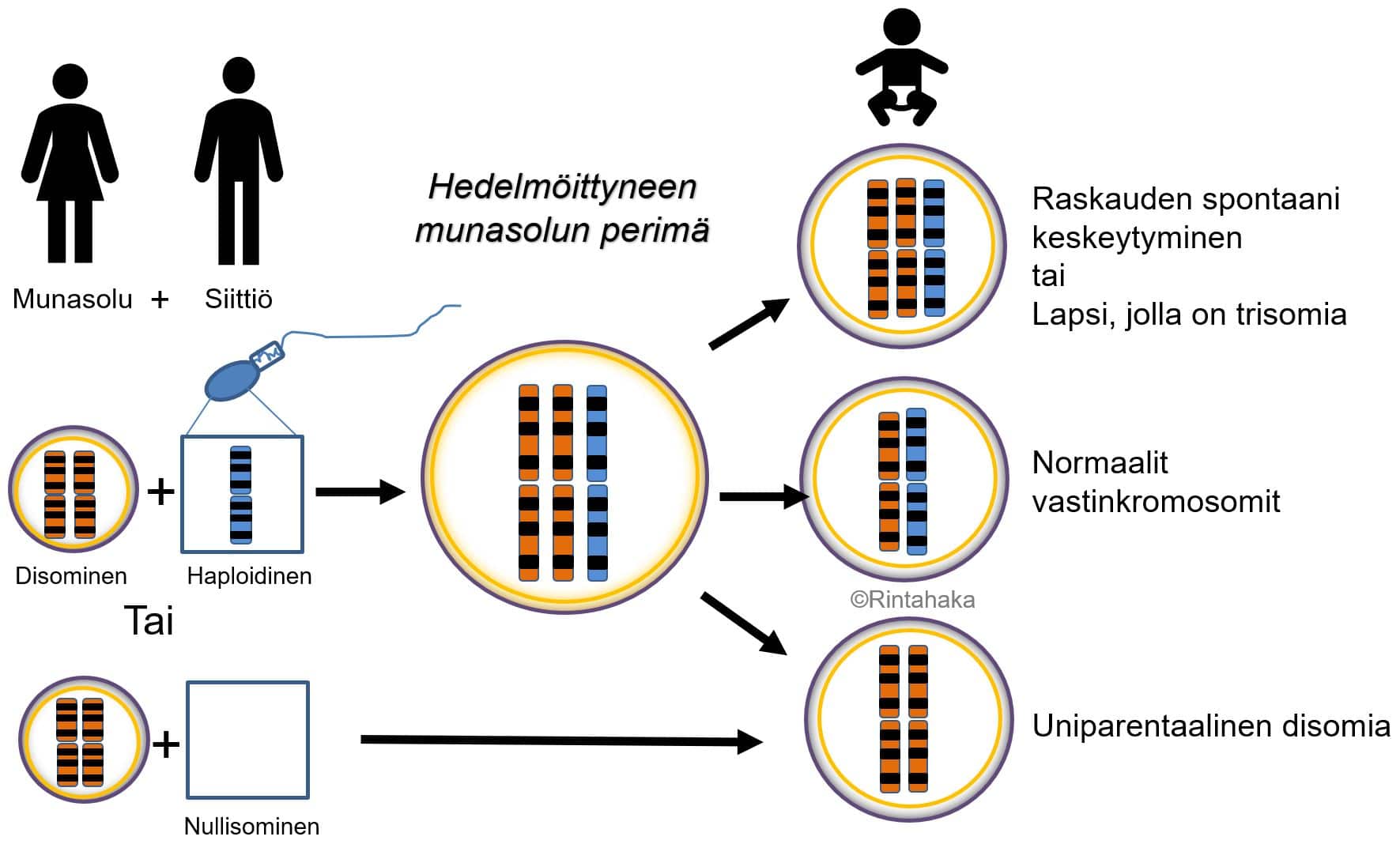
UPD luokitellaan edelleen vanhemmalta perityn uniparentaalisen kromosomin mukaan joko uniparentaaliseksi isodisomiaksi tai heterodisomiaksi. Isodisomiassa molemmat yksilöön jääneet kromosomit ovat samanlaiset, koska ne ovat kopioita toisen vanhemman toisesta vastinkromosomista. Heterodisomiassa yksilön kromosomiparin kromosomit poikkeavat toisistaan, koska ne ovat kopioita toisen vanhemman kromosomiparin kummastakin kromosomista. Heterodisomia käsittää yleensä 66 % ja isodisomia 33 % kaikista UPD-tapauksista.
5.1 Mitä käytännön merkitystä UPD:llä voi olla?
UPD voi kohdistua ihmisen kaikkiin 23:een eri vastinkromosomiin. Useimmissa tapauksissa UPD ei aiheuta haittaa, mutta toisinaan se voi tuoda ilmi lapsessa jonkin autosomissa resessiivisesti eli peittyvästi periytyvän sairauden/oireyhtymän, jonka oireeton kantaja lapsen toinen vanhemmista on. Esimerkiksi suomalaiseen tautiperintöön kuuluva rusto-hiushypoplasia voi periytyä UPD:n kautta.
UPD voi johtaa myös kehitysvammaoireyhtymään geneettisen leimautumisen (engl. imprinting) välityksellä (ks. yllä). Jos molemmat vastinkromosomit peritään poikkeuksellisesti UPD:n vuoksi vain toiselta vanhemmalta ja kyseisen kromosomin jokin tietty geeni onkin leimattu ja siten inaktiivinen, lopputulos on sama kuin geeniä ei olisi perimässä lainkaan. Toisin sanoen leimattuja geenejä ei käytetä, vaikka niistä muuten voisikin valmistua toimivia geenituotteita. Ei siis ole yhdentekevää kummalta vanhemmalta vastinkromosomit peritään UPD:ssä ja vielä, kumman vanhemman geenileimat UPD-kromosomien geeneissä on.
Esimerkkejä oireyhtymistä, jotka voivat johtua UPD:stä ovat Prader-Willin oireyhtymä, jonka aiheuttaa äidiltä peräisin olevan kromosomin-15 tietyn alueen (11q2-q13) UPD; Angelmanin oireyhtymä, jossa UPD kohdistuu isän kromosomiin 15; Russel-Silverin oireyhtymä, jossa kromosomin 7 UPD on äidiltä peräisin ja Beckwith-Wiedemannin oireyhtymä, jonka taustalla on isältä peritty kromosomin 11 UPD. Muchandani-Bhoj-Conlinin oireyhtymä johtuu puolestaan kromosomin 20 maternaalisesta eli äidiltä peräisin olevasta UPD:stä.
Harvinaiskeskus Norio tarjoaa kaikille valtakunnallista ja ilmaista keskustelutukea ja ohjausta perinnöllisyyteen ja harvinaisiin sairauksiin liittyvissä asioissa. Lisää tietoa perinnöllisyyshoitajan palveluistamme löydät osiosta Keskustelutuki ja ohjaus.
Takaisin sisällysluetteloon.
Päivitetty 11/2021, 2/2022, 11/2024 ja 3/2025.