Mulchandani-Bhoj-Conlinin oireyhtymä, (UPD(20)mat)
Mulchandani-Bhoj-Conlinin oireyhtymä on harvinainen kromosomimuutoksesta johtuva oireyhtymä. Sille ominaista on syntymää edeltävä ja syntymän jälkeinen hidas kasvu, iso päänympärys suhteessa muun vartalon kokoon, vaikea syömisvaikeus, jotkin hormonaaliset muutokset ja lyhytkasvuisuus.
Harvinaiskeskus Norio, Johanna Rintahaka, FT., 16.7.2025
ORPHA: 96186
ICD-10: Q99.8
ICD-11: LD45.0
OMIM: 617352
Avainsanat: Mulchandani-Bhoj-Conlin syndrome (MBCS), Maternal uniparental disomy of chromosome 20 syndrome, Maternal UPD(20), UPD(20)mat
Lyhyesti
Muchandani-Bhoj-Conlinin oireyhtymä tunnetaan myös nimeltä kromosomin-20 maternaalinen uniparentaalinen disomia oireyhtymä (Maternal uniparental disomy of chromosome 20 syndrome, UPD(20)mat). Oireyhtymä on harvinainen ja siihen liittyy merkittävä lapsen kasvun hidastuminen jo ennen syntymää, syömisvaikeudet, jotkin ulkonäköpiirteet sekä usein myös lyhytkasvuisuus. Muchandani-Bhoj-Conlinin oireyhtymän oirekuva vaihtelee yksilöllisesti, eikä tässä kuvattuja oireita ole kaikilla henkilöillä, joilla on tämä oireyhtymä. Vastaavasti oirekuvaan voi liittyä myös joitakin muita piirteitä, joita ei tässä kuvauksessa ole mainittu.
Oireet ja löydökset
Vauvalla on imemis- ja syömisvaikeuksia, mikä vaarantaa hoitamattomana hänen kasvunsa ja kehityksen. Imeväisikäiseltä voi puuttua myös tavanomaiselle syömiskäyttäytymiselle tyypillinen nälkäitku. Monilla ravitsemustilanne korjataan nenä-mahaletkun välityksellä tai vatsanpeitteiden läpi vietävän ravitsemusavanteen (PEG, perkutaaninen endoskooppinen gastrostomia) avulla. Monilla ravitsemustuesta voidaan luopua varhaislapsuuden jälkeen. Taaperoikäinen ja sitä vanhempi lapsi saattaa olla ruokahaluton, syödä pieniä ruoka-annoksia ja tarvita kannustusta ja kehotuksia syömistilanteissa. Osalla vastasyntyneistä todetaan hypotonia eli alentunut lihasjänteys, joka osaltaan voi vaikeuttaa syömistä. Hypotonia hellittää usein lapsen varttuessa.
Vastasyntyneen pää näyttää suurelta verrattuna muun vartalon kokoon. Myös otsa voi olla korostunut. Nämä piirteet usein korjaantuvat lapsen kasvun myötä. Lievät kasvojen muut erityispiirteet ovat mahdollisia. Ulkonäköpiirteisiin voivat lukeutua myös skolioosi eli selkärangan sivuttainen vinouma, hyperlordoosi eli notkoselkäisyys ja/tai poikkeavuudet sormissa ja/tai varpaissa. Toisinaan ihossa voi olla pigmenttimuutoksia, kuten maitokahviläiskiä tai voimakkaasti tai epäsäännöllisesti pigmentoituneita alueita. Edellä mainittuja ulkonäköpiirteitä ei kuitenkaan ole kaikilla.
Kehitysviive on mahdollinen, mutta harvinainen Muchandani-Bhoj-Conlinin oireyhtymässä. Se voi ilmetä motoristen eli liikkumisen perustaitojen omaksumisessa tai puheen kehityksessä. Kehitysvamma ei ole tyypillinen oirekuvassa.
Muchandani-Bhoj-Conlinin oireyhtymässä kasvuhormonin puutos voi olla mahdollinen. Tätä tilaa hoidetaan kasvuhormonihoidolla, ja monilla se korjaa kasvupulmia. Viimeaikaisissa tutkimuksissa on viitteitä myös mahdollisista muutoksista lisäkilpirauhashormonin ja tyreotropiinin (TSH) erityksessä ja vasteissa. Hormonimuutokset voivat kehittyä ajansaatossa ja siksi hormonipitoisuuksia on hyvä mitata ja seurata säännöllisesti.
Toisinaan poikalapsilla todetaan laskeutumattomat kivekset.
Syy ja perinnöllisyys
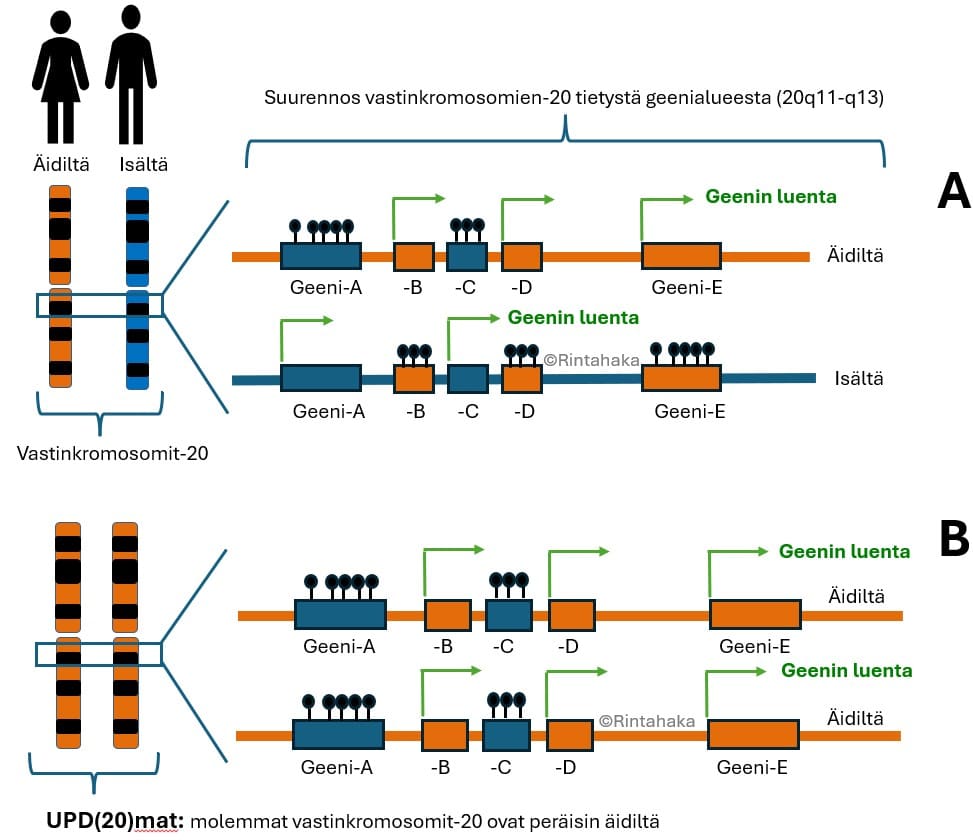
Muchandani-Bhoj-Conlinin oireyhtymässä molemmat kromosomit numero 20 ovat peräisin äidiltä. Ilmiötä, jossa lapsi on perinyt vastinkromosominsa vain toiselta vanhemmaltaan (isältä tai äidiltä) kutsutaan uniparentaaliseksi disomiaksi (UPD). Oireyhtymän oireet johtuvat geeniepätasapainosta, koska kromosomissa-20 on geenejä, joista osaa luetaan vain äidin vastinkromosomista-20 ja osaa vain isän vastinkromosomista-20. Kun Muchandani-Bhoj-Conlinin oireyhtymässä molemmat vastinkromosomit ovat peräisin äidiltä, tietyistä isän vastinkromosomin-20 geeneistä ei siis pystytä valmistamaan geenituotteita, koska isän vastinkromosomia-20 ei ole perimässä. Tästä siis seuraa oireyhtymän oireet.
Uniparentaalisen disomian (UPD) syntymekanismeja on useita, ja ne kaikki johtuvat kromosomien jakautumishäiriöistä. Vanhempien ikääntyminen, etenkin 35-ikävuoden jälkeen, lisää erilaisten kromosomimuutosten todennäköisyyttä. Muchandani-Bhoj-Conlinin oireyhtymää käsittelevissä julkaisuissa äidin ikä on usein ollut yli 40-vuotta. Lisää tietoa uniparentaalisesta disomiasta ja sen syntytavoista voit halutessasi lukea Harvinaiskeskus Norion Syventävää tietoa perimästä-sivuilta kohdasta ”Tunnetuimmat epätyypilliset periytymistavat, Mitä on uniparentaalinen disomia, UPD?”
Perhe voi halutessaan keskustella perinnöllisyysneuvonnassa oireyhtymän toistumistodennäköisyydestä ennen seuraavaa mahdollista raskautta. Harvinaiskeskus Norion sivuilta löytyy tietoa myös perhesuunnittelusta tilanteissa, joissa perheessä on mahdollisesti kohonnut todennäköisyys johonkin harvinaissairauteen: Perhesuunnittelu ja raskaus. Kaikista näistä aiheista voi myös keskustella ilman lähetettä ja veloituksetta Harvinaiskeskus Norion perinnöllisyyshoitajan kanssa. Yhteystiedot keskustelutukeen ja neuvontaan löydät tämän julkaisun lopusta.
Yleisyys
Oireyhtymän yleisyydestä ei ole olemassa luotettavia tietoja. Lääketieteellisessä kirjallisuudessa on julkaisuja noin 20 henkilöstä, joilla on Muchandani-Bhoj-Conlinin oireyhtymä.
Diagnoosi ja hoito
Erotusdiagnostiikassa on huomioitava Russel-Silverin oireyhtymä ja Templen oireyhtymä. Russel-Silverin oireyhtymän taustalla voi olla kromosomin-7 maternaalinen uniparentaalinen disomia (UPD(7)mat) ja Templen oireyhtymä johtuu puolestaan maternaalisesta kromosomin-14 uniparentaalisesta disomiasta (UPD(14)mat). Yhteneväistä Muchandani-Bhoj-Conlinin oireyhtymällä ja Russel-Silverin oireyhtymällä ovat lisäksi mm. merkittävät syömisvaikeudet, alhainen syntymäpaino, kasvun hidastuminen (failure to thrive) sekä jotkin yhteneväiset ulkonäköpiirteet. Muchandani-Bhoj-Conlinin oireyhtymän diagnoosi voidaan poissulkea tai vahvistaa tietynlaisten perimäntutkimusten avulla. Tutkimukset tehdään verinäytteestä, ja vastausten saaminen kestää yleensä pari kuukautta.
Diagnoosin saaminen lopettaa oireiden syiden etsimisen erilaisin tutkimuksin, selkiyttää ennustetta ja auttaa arvioimaan oireyhtymän periytymistodennäköisyyttä perheessä tai lähisuvussa. Nykyisin tiedetään, että monet harvinaiset sairaudet ovat syntyneet niin sanotun uuden (de novo) muutoksen seurauksena, jolloin vanhemmat eivät ole kyseisen perimänmuutoksen kantajia ja siksi sisaruksien riski sairastua on hyvin pieni.
Oireyhtymää ei voida parantaa, mutta sen yksittäisiä oireita pystytään hoitamaan. Hoito on oireiden mukaista. Ravitsemustilanteen korjaaminen on erittäin tärkeää. Tähän voidaan käyttää tarvittaessa nenämahaletkua tai PEG-ravitsemusavannetta. Puheterapiassa voidaan harjoitella syömiseen ja nielemiseen liittyviä taitoja. Fysio- ja toimintaterapian tarve arvioidaan yksilöllisesti. Kasvuhormonin käytöstä on hyviä tuloksia.
Ennuste
Muchandani-Bhoj-Conlinin oireyhtymän ennuste on hyvä.
Historia
Oireyhtymä tunnistettiin ensimmäisen kerran vuonna 2015. Se on saanut nimensä tutkijoiden Urabhi Mulchandani, Elizabeth J. Bho ja Laura K. Conlinin sukunimien mukaan. Oireyhtymää kutsutaan myös maternaaliseksi kromosomin-20 uniparentaaliseksi disomiaksi perimässä tapahtuneen kromosomimuutoksen mukaan.
Kokemustietoa
Onko sinulla omakohtaista kokemusta tästä diagnoosista? Keräämme kokemustietotarinoita, ja sinäkin voit osallistua. Lue lisää Kokemustietoa-sivulta.
Huomioitavaa
Kromosomin-20 paternaalinen uniparentaalinen disomia (UPD(20)pat) johtaa pseudohypoparatyreoosi-1b:hen, jossa lisäkilpirauhashormonin kohdekudokset eivät reagoi lisäkilpirauhashormoniin, vaikka sitä kehossa olisikin. Pseudohypoparatyreoosin oireet ovat samankaltaiset kuin lisäkilpirauhasen vajaatoiminnassa.
Tukipalvelut
Harvinaiskeskus Noriosta voi tiedustella vertaistukea. Lue lisää Vertaistuki-sivultamme.
Harvinaiskeskus Norion perinnöllisyyshoitajaan voi ottaa yhteyttä, kun haluaa keskustella perimään tai harvinaissairauksiin liittyvistä asioista. Lue lisää Keskustelutuki ja neuvonta -sivultamme tai soita 044 5765 439.
Tukiliiton sivuilta löytyy runsaasti tietoa erilaisista palveluista: Tuki ja neuvot.
Facebookista löytyy hakusanalla ”Mulchandani-Bhoj-Conlin Syndrome” oma englanninkielinen keskusteluryhmä läheisille, joita oireyhtymä koskettaa. Jäseneksi ryhmään pääsee pyytämällä ryhmän jäsenyyttä.
Aiheesta muualla
ERN-ITHACA (European Reference Network for Rare Malformation Syndromes, Intellectual and Other Neurodevelopmental Disorders) eli synnynnäisten rakenteellisten poikkeavuuksien ja harvinaisten kehitysvammaoireyhtymien osaamisverkosto
Suomenkielistä lisätietoa ERN-ITHACA:sta löydät Harvinaiskeskus Norion sivuilta kohdasta: ERN-ITHACA, synnynnäisten rakennepoikkeavuuksien ja harvinaisten kehitysvammaoireyhtymien verkosto
Lähteet
Orphanet: Maternal uniparental disomy of chromosome 20 syndrome
Online Mendelian Inheritance in Man (OMIM): Mulchandani-bhoj-conlin syndrome; MBCS
Tannorella, P., Minervino, D., Guzzetti, S., Vimercati, A., Calzari, L., Patti, G., Maghnie, M., Allegri, A. E. M., Milani, D., Scuvera, G., Mariani, M., Modena, P., Selicorni, A., Larizza, L., & Russo, S. (2021). Maternal Uniparental Disomy of Chromosome 20 (UPD(20)mat) as Differential Diagnosis of Silver Russell Syndrome: Identification of Three New Cases. Genes, 12(4), 588. https://doi.org/10.3390/genes12040588
Mulchandani, S., Bhoj, E. J., Luo, M., Powell-Hamilton, N., Jenny, K., Gripp, K. W., … & Conlin, L. K. (2016). Maternal uniparental disomy of chromosome 20: a novel imprinting disorder of growth failure. Genetics in Medicine, 18(4), 309-315. DOI: 10.1038/gim.2015.103
Julkaistu ensimmäisen kerran Harvinaiskeskus Norion sivuilla 16.7.2025